Enfermedades genéticas: características, métodos de diagnóstico y cuadros clínicos
•
0 likes•855 views

1) El documento describe varias enfermedades genéticas, incluyendo su característica genética, método de diagnóstico y cuadro clínico. 2) Algunas de las enfermedades discutidas son deficiencia de 5-alfareductasa, aciduria glutárica, acondroplasia, albinismo, síndrome de Down y distrofia muscular de Duchenne. 3) Los métodos de diagnóstico incluyen exámen físico, pruebas de sangre, electrocardiograma, cariotipo y PCR.

Recommended
More Related Content
What's hot
What's hot (20)
Similar to Enfermedades genéticas: características, métodos de diagnóstico y cuadros clínicos
Similar to Enfermedades genéticas: características, métodos de diagnóstico y cuadros clínicos (20)
More from Facultad de Medicina UANL
More from Facultad de Medicina UANL (20)
Recently uploaded
Recently uploaded (20)
Enfermedades genéticas: características, métodos de diagnóstico y cuadros clínicos
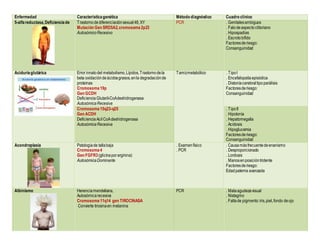
- 1. Enfermedad Característicagenética Método diagnóstico Cuadro clínico 5-alfareductasa,Deficienciade Trastornodediferenciaciónsexual46,XY Mutación Gen SRD5A2,cromosoma2p23 AutosómicoRecesivo PCR . Genitalesambiguos . Falodeaspecto clitoriano . Hipospadias . Escrotobífido Factoresderiesgo: Consanguinidad Aciduria glutárica Error innatodel metabolismo,Lípidos,Trastornodela beta oxidacióndeácidosgrasos,enla degradaciónde proteínas Cromosoma19p Gen GCDH Deficiencia Glutaril-CoAdeshidrogenasa AutosómicaRecesiva Tamizmetabólico . Tipol . Encefalopatíaepisódica . Distoníacerebraltipoparálisis Factoresderiesgo: Consanguinidad Cromosoma15q23-q25 Gen ACDH Deficiencia Acil CoAdeshidrogenasa AutosómicaRecesiva . Tipoll . Hipotonía . Hepatomegalia . Acidosis . Hipoglucemia Factoresderiesgo: Consanguinidad Acondroplasia Patologíade tallabaja Cromosoma4 Gen FGFR3 (glicinaporarginina) AutosómicaDominante . Examenfísico . PCR . Causamásfrecuentedeenanismo . Desproporcionado . Lordosis . Manosenposicióntridente Factoresderiesgo: Edadpaterna avanzada Albinismo Herenciamendeliana, Autosómicarecesiva Cromosoma11q14 gen TIROCINASA Convierte tirosinaen melanina PCR . Malaagudezavisual . Nistagmo . Faltade pigmento:iris,piel,fondo deojo
- 2. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Alcohol-fetal,Síndromede Trastornodelneurodesarrollo Anamnesisa la madre . Microcefalia . Ausenciadefiltrum . Implantaciónbajadeorejas Factoresderiesgo: Ingesta de alcohol Alzheimer,Enfermedad de Herenciamultifactorialdeladulto,noha sidoidentificado un genespecífico. Mutación deloscromosomas1,14 y 21 Cromosoma19Gen ApoE2,ApoEyApoE4 . Proteína precursoradeamiloidebeta . GenApoE (apolipoproteínaE) --- despuésde los5O años . Genpsen2(presenilina2) --- antesde los 5O años . Depósitosamiloidesen ovillos neurofibrilaresyen placasneuronales . Causamásfrecuentededemencia . Deterioroprogresivoe irreversibledel intelecto, memoriayhabilidadessocialesyemocionales Factoresderiesgo: . Traumatismocraneoencefálico . Ictus . SíndromedeDown* . SIDA Anemiadecélulasfalciformes Herenciamendeliana, Autosómicarecesiva Cromosoma11 Gen Betaglobina Frotissanguíneo PCR . Debilidadycansancio . Microinfartos . Insuficienciacardiacayrenal Angelman,Síndromede DisomíauniparentalpaternaoImprontagenómica materna Cromosoma15q11.2 ladeleciónsedetectaconFISH Gen UBE3A(UbiquitinProteinLigasaE3A) Se activa UBE3A y se inactivaSNPRN Hay unamicrodeleciónmaterna(sinohubieradeleciónno habríaSA ya quese sabe queen esta regiónde cromosomasolamenteseexpresaellocimaterno),con Disomíapaterna(ya queel alelopaternosustituye esta deleción) PCR metilada FISH . Ausenciadelalelomaterno . Epilepsia . Retrasomentalgrave . Marchaatáxica . Aspecto feliz . Caminaconlosbrazoslevantados Microcefalia Hablainexistenteo limitada Trastornosdelsueño Risay excitabilidadconstantes
- 3. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Ataxiade Friedereich HerenciaAutosómicaRecesiva, Expansióndetripletes GAA Cromosoma9Gen FXN:Frataxina PCR . Deterioroprogresivode cerebelo . Pérdidadesensibilidad . Descoordinacióndemovimientos . Disfagia,disartria Atrofiamuscularespinal Herenciamendeliana, Autosómicarecesiva Cromosoma5q13 gen SMN1 Electromiografía PCR . Degeneracióndelascélulasdelasastas anterioresde lamédulaespinal,queproduce debilidadmuscularprogresivay muerte. BardetBiedl,Síndromede Herenciaoligogénica Cromosomas2,3, 4, 7, 9, 11, 12, 14, 15, 16, 17, 20 Herenciatrialélica:Unindividuoeshomocigotopara mutacionesen unlocus, pero tambiénheterocigotopara unamutación enotro locus . Pigmentaciónretiniana . Anosmia . Anomalíasrenales . Es multisistémica,Principalmentese caracterizapormanifestacionesde: . Obesidad . Retinitispigmentosa . Polidactilia . Retrasomental . Hipogonadismo Becker,Distrofiamuscularde Herenciamendeliana, Recesiva ligadaalcromosomaX CromosomaXp21 GenDMD Signode Gowers Electromiografía PCR . Debilidadprogresiva . Ataxia . Lordosis . Signode Gowers(apoyarmanosen rodillas) . Insuficienciacardiorrespiratoria
- 4. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Beckwith Wiedemann,Síndrome de DisomíauniparentalpaternaoImprontagenómica materna Cromosoma11p15.5 Gen IGF2 (Factordecrecimientodelainsulina2) Insulin- likegrowth factor 2 . HipermetilacióndeDMR1(IGF2estimulada) PCR metilada . Ausenciade alelo materno . Crecimientoexcesivo . Defectosenparedabdominal . HIPOGLICEMIAneonatal . TumordeWilms . Hemihiperplasia,visceromegalia,anomalías renales,auriculares,paladarhendido . Macrosomía(crecimientoenexcesoprenatal y/o posnatal) . Macroglosia(lenguagrande) . Defectosdela paredabdominal(onfalocele, herniaumbilical,diastasisrectal) Cáncercolorrectal sin poliposis hereditario (SíndromedeLynch) CáncerColorrectal Cromosoma 3p21.3 GenesMSH2,MLH1 enel90%de los casos MSH6en el 7 al 10% hPMS2en el5% Estos genes participanenlareparacióndeloserroresque se producendurantelareplicacióndelADN AutosómicaDominante Tx.Quimioterapia . Estreñimientoodiarreapersistentes . Dolorabdominal . Fatiga . Hecessanguinolientas Factoresderiesgo: . Mutación . Alcohol . Consumoexcesivodegrasas Cáncerdemamafamiliar Cromosoma13q13.1 Gen BRCA2 Mamografía PCR . Edadtempranadeaparición . Familiaresdeprimergrado Cáncerdemama-ovárico- familiar Cromosoma17q21 Gen BRCA1 Cáncerdemama-ovarioESPORÁDICO, Cromosoma11 gen EMSY Mamografía Ecografíadoppler PCR . Tumoraciónenmama . Enrojecimiento . Hinchazón . Retracciónpezón Factoresderiesgo: . NO AMAMANTAR . Nohaber tenidohijos . Menarquíatemprana . PenetranciadeBRCA: 8O%
- 5. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Cardiopatíascongénitas Herenciamultifactorialcongénita Gen TBX1y PTPN11 (tirosin-fosfatasaSHP-2 proteínade señalización) Gen NKX2.5y GATA4 Mutaciones específicasen cada uno de estos genesrepresentan severas anormalidades cardiacascomo: Defectos septales (GATA4 del cromosoma 8) Defectos del septum atrioventricular (CSX/NKX2.5 del cromosoma 5) Defectos en la conducción, estenosis pulmonar(Síndrome de Noonan y LEOPARD, cromosoma11) y Síndrome de DiGeorge y velo caridiofacial (TBX1 Cromosoma 22) . Exploraciónfísica . Electrocardiograma . Defectosestructurales(detabicación,válvulas, etc.), vasculares. Máscomúnestenosisaortica. Factoresderiesgo: - Enfermedadesgenéticas Cri-du-Chat,Síndromede Cromosomopatíaestructural, (DELECIÓN5p15) Mutacióndenovo 85% Progenitorc/translocación 15% Cariotipo FISH . Retrasode crecimientoymental . Llantode tonoalto maldesarrollo laríngeo . Microcefalia . Carade lunallena . Hipertelorismo . Orejasdeimplantaciónbaja . Plieguesimiesco . Anomalíascardiacasygenitourinarias Defectosdel tubo neural Herencia multifactorialcongénita Cromosoma1p36.3Gen MTHFR, (MetilenoTetraHidroFolatoReductasa) Polimorfismo677C> T Examenfísico PCR Abiertas . AnencefaliaMielomeningocele Cerradas . Espinabífida, encefalocele. Factoresderiesgo: . Embriopatíaporácidovalproico . Deficienciadeácidofólico Deleción 1p36 Cromosomopatíaestructural, Síndromedegenescontiguos (MICRODELECIÓN1p36) FISH . Retrasomentaly deldesarrollo . Hipotonía . Epilepsia . Sorderay problemasvisuales . Cejasrectas . Cardiomiopatías
- 6. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Diabetesmellitus Herenciamultifactorialdeladulto Cromosoma6 Gen HLADR3/DR4 (antígenos leucocitarios humanos HLA confoman el complejo mayor de histocompatibilidad MHC) . Químicasanguínea TipoI Factoresderiesgo: Confiereriesgode SIRENOMELIA LociINSVNTRpredisponeaDM l Herenciamultifactorialdeladulto Cromosoma2 Gen CAPN1O (Calpaína10en Mexico) Cromosoma10 Gen TCF7L2 . Químicasanguínea TipoII Factoresderiesgo: Obesidad Herenciamultifactorialdeladulto GenesHNF1alfa,HNF1beta,HNF-4alfa,IPF1alfay NEUROD1)yla glucocinasahepática(GKC) Glucosinasaformaleve Fenotipo Gen Proteína MODY1 HNF4A HNF-4alfa MODY2 GKC Glucosinasa MODY3 HNF1A HNF-1alfa MODY4 IPF1 IPF-1 MODY5 HNF1B HNF-1beta . Químicasanguínea TxMODY--- SULFONILUREA Mody(diabetesdela madurezde inicioenla juventud) Factoresderiesgo: Antecedentesfamiliares DiGeorge,Síndromede Cromosomopatíaestructural, Síndromedegenescontiguos MICRODELECIÓN22q11 Haploinsuficienciadel gen TBX1 FISH . Retrasomental . Micrognatia . Paladarhendido . Hipoplasiaparatiroidea(Hipocalciemia) . Hipoplasiatímica(Infeccionesrecurrentes) . TetralogíadeFallot . Esquizofreniaen adultez DisgenesiagonadalXY Trastornodediferenciaciónsexual46,XY CromosomaYp11.3 Mutación en el gen SRY AutosómicaDominante,De novo PCR . Gónadasrudimentarias . Hipogonadismohipergonadotrófico . Amenorreaprimaria . Úteroy trompashipoplásicos Factoresderiesgo: Antecedentesfamiliares DisgenesiagonadalXX Trastornodediferenciaciónsexual46,XX Cromosoma2p21 Mutaciónen el gen Receptordela FSH, Mutaciónde novo PCR . Fenotipofemenino . Genitalesinternosyexternos femeninos . Infantilismosexual . ConcentracioneselevadasdeFSHy LH Factoresderiesgo: Sexo femenino
- 7. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Displasiacongénitadecadera Herenciamultifactorialcongénita Examenfísico . Luxacióndelacabezadelfémurdel acetábulo Factoresderiesgo: . Mayorfrecuenciaensexofemenino Distrofiamiotónica HerenciaAutosómicaDominante, Expansión detripletesCTG Cromosoma19 Gen DMPK Electromiografía PCR . Hipotonía . Defectosdeconduccióncardiaca . Alteracióndeperistalsis . Cálculosbiliares . DiabetesMellitustipo1 . Calviciefrontal Down,Síndromede Cromosomopatíanumérica, Aneuploidía(TRISOMÍA21) 47 XY+21/ Trisomía 95% Translocaciónrobertsoniana 4% Mosaicismo 1% Cariotipo Análisis de vellosidades coriónicas Cribadoprenatal Prueba“Triples”(disminuye alfa-feto, estriol, aumenta hCG) . Hipotoníagrave . Braquicefalia . Hendiduraspalpebralesinclinadashaciaarriba . Orejaspequeñas . Lenguaprotruyente (Macroglosia) . Clinodactiliadequintafalange . Líneapalmarúnica(plieguesimiesco) . Defectoscardiacos . Atresia anal o duodenal(ecografíasignodoble burbuja) NeoplasiamáscomúnLeucemialinfoblástica aguda Duchenne,Distrofiamuscularde Herenciamendeliana, Recesiva ligadaalcromosomaX CromosomaXp21 GenDMD Signode Gowers Electromiografía PCR . Debilidadprogresiva . Ataxia . Lordosis . Signode Gowers(apoyarmanosen rodillas) . Insuficienciacardio-respiratoria
- 8. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Edwards,Síndromede Cromosomopatíanumérica, Aneuploidía(TRISOMÍA18) Ultrasonido Cariotipo . Retrasode crecimiento . Retrasomentalgrave . Microcefalia . Occipitalprominente . Orejasdisplásicasdeimplantaciónbaja . Micrognatia . Panículoadiposoymasamuscularescasa . Cardiopatíacongénita . Anomalíasgastrointestinales(divertículode Meckel) . Anomalíasgenitourinarias(riñónenherradura) . Manostrisómicas(puñofuertementecerrado) . Uñasde manosy pieshipoplásicas . Talónprominente . Piesen mecedora EhlersDanlos,Síndromede Herenciamendeliana, Autosómicadominante Cromosoma17q21 Gen COLAGENATIPO III PCR . Pielhiperelástica . Articulacionesmuylaxas(dislocaciones) . Prolapsodeválvula mitral . Fácildañodevasos sanguíneos - Def. Lisil-Hidroxilasa Enfermedad coronaria Herenciamultifactorialdeladulto Gen MEF Gen HMG-CoA Electrocardiograma . Aterosclerosisdearterias coronarias . Angina depecho Factoresderiesgo: . Tabaquismo . Sedentarismo Epidermólisisbullosa Herenciamendeliana, Autosómicarecesiva Cromosoma 3p21Gen ColágenaVII PCR . Alopecia . Ampollasy erosionesenpiely mucosa
- 9. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Esclerosistuberosa Herenciamendeliana, Autosómicadominante Cromosoma9 GenTSC1 Cromosoma16 GenTSC1 PCR . TríadadeVogt: Angiofibromas- Epilepsia - Retrasomental . Tuberosidades:(causaderetrasomental) tejidoen formade tubérculoencerebro . CalcificacionesenelSNC(causade epilepsia) Fenilcetonuria Error innatodel metabolismo,Aminoácidos DeficienciaFENILALANINAHIDROXILASA AutosómicaRecesiva Cofactor-Tetrahidrobiopterina Dx --- HPLC (High PerformanceLiquid Chromatography)para diferenciarentre HIPERFENILALANINEMIA & PKU. Orinaolora moho . Retrasomental . Hipopigmentación . Epilepsia Altos niveles ácido fenilpirúvico Factoresderiesgo: Consanguinidad Fibrosisquística Herenciamendeliana, Autosómicarecesiva Cromosoma7q31 Gen CFTR) PCR Cloruroen sudorGold Standar . Acumulacióndesecrecionesespesas . Insuficienciapancreática . Diabetesmellitus . EPOC . Ictericia(obstrucciónvías biliares) . Varones estériles(agenesiaconductos deferentes) Galactosemia Error innatodel metabolismodeCarbohidratos DeficienciaGALACTOSA 1-FOSFATO URIDIL TRANSFERASA (GALT) AutosómicaRecesiva Tamizmetabólico Tx.Lechedesoya LACTANTE . Rechazodel alimento . Hepatomegalia . Letargo . Vómito Factorderiesgo: consanguinidad JOVEN . Cataratas . Retrasomental . Cirrosis Factorderiesgo: consanguinidad
- 10. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico HemofiliaAy B Herenciamendeliana,Recesivaligadaal cromosomaX A (gen FACTOR8cromosomaXq28) B (gen FACTOR9cromosomaXq28) Tiemposde sangrado, coagulaciónyprotrombina PCR . Hemorragiasenarticulacionesquesoportan peso . Artropatía progresiva Hijo demadrediabética Patologíade tallaalta . Examenfísicoy de laboratorio . HIPOGLUCEMIAalnacimiento . Macrosomía . Aumentode peso Bebe grande Factoresderiesgo: . Diabetesgestacional=Producto macrosómico SIN dismorfias . Diabetespregestacional=Producto macrosómicoCONdismorfias Hipercolesterolemiafamiliar Herenciamendeliana, Autosómicadominante Cromosoma19 Gen LDLR PCR . Coronariopatíaprematura . Xantomas(depósitosubcutáneodelípidos) . Aterosclerosis . Infartos . Receptor deLDLafectado Hiperplasiasuprarrenal congénita Trastornodediferenciaciónsexual46,XX Deficiencia21ALFA HIDROXILASA AutosómicaRecesiva TestdeACTH . Cariotipodemujer;Fenotipodevarón . Hirsutismo Factoresderiesgo: Consanguinidad HipomelanosisdeIto Mosaicismo Translocación del Xp11 Frecuenciadetrastornoneurocutáneos: Neurofibromatosis Esclerosistuberosa HipomelanosisdeIto Examenfísico . Retrasomental . Convulsiones . Lesionesenpieltipo máculas,patróndelíneas de Blaschko
- 11. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Hipoxiaperinatal Trastornodelneurodesarrollo Examenfísico Retrasomental Factoresderiesgo: Manejoincorrectodelparto Hunter,Mucopolisacaridosisde Error innatodel metabolismo,Carbohidratos, MucopolisacaridosisII CromosomaXDeficienciaIDURONATO SULFATO SULFATASA Tamizmetabólico . Retrasomental . Macroglosia . Hepatoesplenomegalia . Anomalíasesqueléticas Factoresderiesgo: . Sexomasculino Huntington,Enfermedad de HerenciaAutosómicaDominante, Expansióndetripletes CAG >40 penetrancia <25 normal Cromosoma4Gen Huntingtina (HTT o IT15) PCR . Corea(movimientosinvoluntariosybruscos) . Deteriorodememoria . Ansiedad, cambiosdehumor . Depresión,paranoia . Insuficienciacardio-respiratoria Hurler,Mucopolisacaridosisde Error innatodel metabolismo,Carbohidratos, MucopolisacaridosisI Cromosoma4 DeficienciaALFA-L-IDURONIDASA (IDUA) GAG depositadoentejidosson el heparánsulfato(HS) y Dermatánsulfato(DS) AutosómicaRecesiva Tamizmetabólico . Orina:excreciónde dermatány heparánsulfato . Retrasomental . Opacidadcorneal . Hepatoesplenomegalia . Anomalíasesqueléticas Factoresderiesgo: Consanguinidad IctiosisligadaaX Herenciamendeliana, Recesivaligadaal cromosomaX gen STS:SULFATASAESTEROIDEA PCR . Hiperqueratosis . Atrofia de ladermis . Pielescamosa(comoladeunpez) . Aspecto Sucio . Loshombressonlos afectados . mujeresportadoraspuedentenerpoca afectación
- 12. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Incontinentiapigmenti Herenciamendeliana, Dominanteligadaal cromosomaX Gen NEMO ModuladoresencialdelNF KappaB. También se le conocecomoIKKoIKBKG. Geninvolucradoenlatransduccióndeseñal PCR . Retrasomental . Convulsiones . Alopeciaareata . Loshombresnosobreviven . Pigmentaciónanormal depiel(Líneasde Blaschko) Insensibilidad alosandrógenos Trastornodediferenciaciónsexual46,XY .MutaciónengenReceptordeandrógenos,cromosomaX Ligadaa X . Tx.Removertestículosno descendidosyadministrar estrógenos . Cariotipodevarón; Fenotipodemujer . Genitalesexternosfemeninos . Ausenciadeútero y trompasde Falopio . Amenorreaprimaria . Desarrollomamario Factoresderiesgo: Sexo masculino Klinefelter,Síndromede Cromosomopatíanumérica, Aneuploidíade cromosomassexuales(TRISOMÍAX) A mayor cantidaddeX, mayor retraso mental Cariotipo . Ligeroretrasomental . Tallaalta . Desproporcióndetroncoyextremidades . Distribucióngrasaginecoide . Hipogonadismo . Azoospermia(infértiles) Labio-paladarhendido Herenciamultifactorialcongénita Examenfísico . Hendidurapequeñaenlabioy/o paladarhasta fisura completa . Frecuenteunilateralderecho Factoresderiesgo: . Anomalíascongénitas . Fármacosanticonvulsivos
- 13. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Leber,Atrofiaópticade Herenciamitocondrial Mitocondria,GenND4 LáminasdeSnellen . Pérdidadela agudezavisual Leucemiamieloidecrónica Translocación cromosómica t(9;22)(q34;q11)que formael cromosomaFiladelfia:BCR-ABLo PHque tieneactividadtirosinacinasa Cáncer hematolinfático, anomalía genética adquirida. A causa de esta translocación se producen 2 nuevosgeneshíbridos:el BCR-ABL (Ph) en el cromosoma 22q- yel gen recíproco ABL-BCRen el cromosoma derivado 9q+,el cual no parece desempeñarninguna actividad funcional en la enfermedad. BiometríaHemática FISH o PCR Tx.Inhibidoresdetirosina cinasa . Leucocitosis . Hiperviscosidaddelasangre . Hipoxiatisular . Esplenomegalia Factoresderiesgo: Causasdesconocidas Grandesdosisde radiación Li-Fraumeni,Síndromede Cáncer Cromosoma17 Mutacióndel gen P53 AutosómicaDominante El Gen P53, El guardián del genoma, es un gen supresor de tumoresque se encuentra con frecuencia mutado en las célulasque componen lostumores malignos. Su función es codificar la proteína p53, la cual impide realizar la división celular si la molécula de ADN no se ha replicado de forma correcta. Debido a ello las mutaciones que afectan a la función del gen hacen al individuo muy propensoal desarrollo del cáncer. PCR . Neoplasiasmalignas:carcinomas,sarcomas, leucemias Factoresderiesgo: Mutaciones LinfomadeBurkitt Cáncerhematolinfático Translocacióncromosómica t(8;14)(q24;q32) Gen MYC Gen MYC tiene la función natural de control en el crecimiento y proliferación celular. Biometríahemática FISH . Adenopatía cervicaleinguinal . Tumoraciónmandibular Factoresderiesgo: . InfecciónconvirusdeEpstein Barr . InfecciónconVIH Marfán,Síndromede Patologíade tallaalta Gen FBN1 (Fibrilina1) AutosómicaDominante Este gen juega un papel importante como pilar fundamental para el tejido conectivo del organismo. Examenfísico . Subluxacióndelcristalino . Pectum excavatum . Articulacioneslaxas . Aracnodactilia . Dilatacióndelaaortaascendente Factoresderiesgo: Antecedentesfamiliares
- 14. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico MELAS,Síndromede (miopatía, encefalopatía, acidosis láctica y episodios semejantes a apoplejías) Herenciamitocondrial 80%de los casosse debena lamutación3243A>G en el Gen del tARNleu (paraleucina) Es muydifícilel asesoramientogenéticodebidoala heteroplasmia Electroencefalografía Electromiografía Resonanciamagnética nuclearcerebral Tomografíacomputarizada Cuadroclínico . Encefalopatía . Epilepsia . Acidosisláctica . Pronóstico desfavorable . Episodiosdeapoplejía . Deterioromental Pérdidade visión y audición . Miopatíasgraves. MERRF,Síndromede (El síndrome de epilepsia mioclónica asociada a fibras rojas rasgadas. Herenciamitocondrial Gen tRNAlys(paralisina) 80 a 90%mutaciónA8344G (transición de adeninaaguanina enla posición 8344delgen) tRNAleu, tRNAlys, tRNAile, tRNAgly Electroencefalografía Electromiografía . Epilepsiamioclónica . Miopatía . Ataxia Neurofibromatosis Herenciamendeliana, Autosómicadominante Gen NF1cromosoma17 Gen NF2cromosoma22 Neurofibromina:supresoradetumores PCR . Manchascaféconleche . Tumoracionescarnosas(neurofibromas) . NódulosdeLisch . Pecasaxilaresoinguinales - Penetrancia completaenadultez Noonan,Síndromede Patologíade tallabaja Cromosoma12q22 Gen ProteínaTirosinafosfatasa,tipo no receptor,11 (PTPN11) AutosómicaDominante Una mutación en el gen PTPN11 es la responsable de un 50% de los individuos afectados; El gen SOS1mutado, del 13% El gen RAF1, del 3-7% y El gen KRAS, de menos del 5% . Los genes NRAS, BRAF y MAP2K1 1% Es similaralTurner,peroel Sx de NoonanSí se hereda. . Bajaestatura . Cuelloalado . Cardiopatíacongénita(estenosispulmonar) . Deformidaddeltórax . Retardoen lapubertad . Ojosde baseampliaoinclinadoshaciaabajo . Orejasdeimplantaciónbaja . Párpadoscaidos(ptosis) . Penepequeño . Criptorquidia Factoresderiesgo: Antecedentesfamiliares
- 15. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Obesidad Herenciamultifactorialdeladulto Genes: LEP, LEPR,POMC,FTO, Gen de la Leptina (LEP)en el cromosoma 7q31.3 Gen receptor de la leptina (LEPR) en el cromosoma 1p31.3 Gen de la Propiomelanocortina (POMC)del cromosoma 2p23.3, está relacionado con los mecanismos de control de la ingesta y Saciedad. Gen FTO asociado a la masa grasa y obesidad 16q12.2 Examenfísico . Hipertrofiadeltejidoadiposo . Cansancio Factoresderiesgo: Confiereriesgopara ESPINA BÍFIDA & DIABETES MELLITUSTIPO ll . Sedentarismo . Ingestaalta de grasas Orinacon olorajarabedearce, Enfermedad de Error innatodel metabolismo,Aminoácidos DeficienciaDESCARBOXILASADELOSBETA- CETOÁCIDOSRAMIFICADOS AutosómicaRecesiva Tamizmetabólico . Hipotoníae hipertonía . Retrasomental . Vómito . Letargoy Coma Factoresderiesgo: Consanguinidad Ornitín transcarbamilasa, deficienciade Error innatodel metabolismo,Aminoácidos,Ciclodela urea CromosomaX,deficiencia ORNITINACARBAMIL TRANSFERASA Tamizmetabólico Restricciónproteínas . Hiperamoniemia . Intoxicaciónaguda . Alcalosis . Muerte . Elevado ácidoorótico Factoresderiesgo: Sexo masculino Osteogénesisimperfecta Herenciamendeliana, Autosómicadominante Cromosoma17q GenCOL1A1y Cromosoma7COL1A2 (COLAGENATIPO I) En la OI tipo I se produce muy poco colágeno, pero de calidad normal. En el otro tipo, el colágeno es de mala calidad estructural, mientras que la cantidad puede también estar reducida. PCR . Huesosy dientesquebradizos . Escoliosis . Pérdidadeaudición .TipoI Escleróticaazul(Leve) . TipoII Fetal(Letal) . TipoIII esclerótica gris(Severa) - TipoIV (Moderada)
- 16. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Parkinson,Enfermedad de Herenciamultifactorialdeladulto Cromosoma 4q21-q22 Gen PARK1 DeficienciadeDopa Examenfísico . Segundodesordenneurodegenerativomás frecuente . Temblorenreposo . Bradicinesia . Inestabilidadpostural Factoresderiesgo: . Traumatismocraneoencefálico . Ictus Patau,Síndromede Cromosomopatíanumérica, Aneuploidía (TRISOMÍA13) Por tratarse de un cromosomaacrocéntrico,puede presentartranslocaciónrobertsoniana Ultrasonido Cariotipo . ANOMALIAS DE LAS ESTRUCTURAS DE LA LÍNEA MEDIA . Retrasode crecimiento . Retrasomentalgrave . Microcefalia . Holoprosencefalia . Frenteaplanada . Anomalíasoculares(microftalmia) . Hendiduralabialypalatal . Micrognatia . Cardiopatía congénita . Anomalíasgenitourinarias(riñónpoliquístico) . Polidactilia Poliposisadenomatosafamiliar CáncerColorrectal Deleción 5q31,Gen APC (supresortumoral) AutosómicaDominante Tx.Colectomía . Póliposadenomatososintestinales(tumores benignos) Factoresderiesgo: PenetranciadeAPC: 9O% PolisomíaX,Síndromede Cromosomopatíanumérica, Aneuploidíade cromosomassexuales(TRISOMÍAX) Cariotipo . Afectaciónligeradeaprendizaje . Fertilidadnormal
- 17. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico PolisomíaY,Síndrome Cromosomopatíanumérica, Aneuploidíade cromosomassexuales(TRISOMÍAY) Cariotipo . Afectaciónligeradeaprendizaje . Fertilidadnormal Pompe,Enfermedad de Error innatodel metabolismo,Carbohidratos, GlucogenosistipoII DeficienciaGLUCOSIDASA-ALFA-1,4LISOSÓMICA AutosómicaRecesiva Tamizmetabólico . AfectaciónMUSCULAR . Hipotonía . Debilidadmuscular . Insuficienciacardiaca . Insuficienciarespiratoria . Hepatomegalia . Cardiomegalia Factoresderiesgo: Consanguinidad. Prader-Willi,Síndromede DisomíauniparentalmaternaoImprontagenómica paterna (genSNRPN (polipéptidoNderibonucleoproteínas nuclearespequeñas)Smallnuclearribonucleoprotein polypeptide Deleción cromosoma15q11 Se activa SNPRNy se inactivaUBE3A Hay unamicrodeleciónpaterna(sinohubieradeleciónno habríaSPW ya que se sabeque enesta regiónde cromosomasolamenteseexpresaellocipaterno),con Disomíamaterna(ya queel alelomaternosustituye esta deleción) PCR metilada FISH . Ausenciadelalelopaterno . Ligeroretrasomental . Hipotonía . Estatura corta . Obesidad . Ojosalmendrados . ImpulsivoCompulsivo . Hipogonadismo Dificultadesdealimentaciónenlafase posnatal Hiperfagiay obesidadenlainfancia Esterilidad Dismorfia Raquitismo hipofosfatémico familiar Herenciamendeliana, Dominanteligadaal cromosomaX Gen PHEX Cuando el PHEXno codifica correctamente a la metalproteasa se produce, en primer lugar, una alteración directa de la mineralización del hueso y seproduce un incremento de los niveles de fosfatonina que provoca un incremento de fosfaturia que da lugar a hipofosfatemia. PCR . Arqueamientodehuesoslargos . Disminuciónde calcitriol . Reabsorcióndeficientedefosfatos enTCP
- 18. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Retinitispigmentosa Herenciaoligogénica La retinitispigmentariaesun conjuntode enfermedades ocularescrónicas Genesmutadoslosresponsablesderetinitis pigmentosaautosómicadominante: - RHO 25 al 30% - RP1 5 al 10 % - RDS 5 al 10% La retinitis pigmentosa es un conjunto de enfermedades oculares crónicas de origen genético y carácter degenerativo Campometría . Cegueranocturna . Visión en túnel . Ceguera . Iniciaenla infanciaoadolescenciayes bilateral. . Disminucióndeagudezavisual . Fotopsias(destellosdeluz) . Dificultaddepercepcióndecolores(etapa avanzada) Retinoblastoma Cáncer Gen RB1, cromosoma13q14 AutosómicaDominante El Gen RB1 su función es la inhibición de la progresión del ciclo celular antes de la entrada en mitosis, de manera que la célula no entra en división hasta que está preparada para ello y se dan las condiciones adecuadas: pRb impide por tanto la proliferación celular. PCR Tx.Temprano: quimioterapia Tardío:removerglobo ocular : Leucocoria .Tumorocularmaligno Factoresderiesgo: Cuandoes hereditario,teconfiereriesgopara desarrollarotroscánceres Rett, Síndromede Trastornodelneurodesarrollo CromosomaXq28,Mutación Gen MeCP2(proteínade unión ametilCpG) PCR . Mayorfrecuenciaenmujeres . AfectacióndelSNC . Crisisconvulsivas . Problemasrespiratoriosqueempeoranconel estrés. . Brazos y piernasflácidos . Discapacidadesintelectualesydificultadesde aprendizaje . Convulsiones . Pérdidadepatronesnormalesdesueño . Problemasgravesen eldesarrollodellenguaje Factoresderiesgo: Sexo femenino Rubéola Trastornodelneurodesarrollo Infecciónperinatal Examenfísico . Cataratas . Sordera . Cardiopatías Factoresderiesgo: . ConfiereriesgoparaDIABETES MELLITUS TIPO I, pues el virus se alojaen páncreasyojo.
- 19. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Silver-Russell,Síndromede DisomíauniparentalmaternaoImprontagenómica paterna Cromosoma11p15.5 . HipometilacióndeH19 (IGF2inhibida) Si no hubieradeleciónnohabríaSRS ya quese sabe que en esta regióndecromosomasolamenteseexpresael locipaterno(elalelomaternosustituye esta deleción) La hipometilacióndeH19conduceaunaIGF2inhibida PCR metilada . Ausenciadealelopaterno . Retrasode crecimiento . Asimetría corporal . Caratriangular,aspectopseudohidrocefálico . Micrognatia Smith-Magenis,Síndromede Cromosomopatíaestructural, Síndromedegenescontiguos MICRODELECIÓN17p11 Cariotipo FISH . Déficitauditivo . Patróndel sueñoalterado . Se producendañoasí mismos . Se abrazan a sí mismos Se metencosasen orificios . Cardiopatías . Escoliosis Sotos,Síndromede Patologíade tallaalta Gen NSD1, Haploinsuficienciaen receptornuclear NSD1 De novo, AutosómicaDominante PCR . Macrocefalia . Dilataciónventricularcerebral . Frentealta . Barbillay narizpuntiagudas . Crecimiento excesivo . Edadósea avanzada . Dismorfias . Fisuraspalpebralesdisminuidas Factoresderiesgo: Antecedentesfamiliares Turner,Síndromede Cromosomopatíanumérica, Aneuploidíadecromosomassexuales(MONOSOMÍA X) Monosomía45,X 50% Mosaico45,X/46,XX 20% (probabilidadde fertilidad) Isocromosoma46,X(Xq) 15% HaploinsuficienciadelgenSHOX(estaturabaja) Ultrasonido Cariotipo . EDEMAal nacimiento(hidropesía) . NO hay retraso mental . Tallabaja . Cuelloalado . Líneade implantacióndecabellobaja . Defectoscardiacos(soplos)coartacióndela aorta . Agenesiagonadal(infertilidad) . Cúbitovalgo . AusenciadecorpúsculodeBarr
- 20. Enfermedad Característicagenética Métododiagnóstico Cuadroclínico Von Gierke,Enfermedad de Error innatodel metabolismodeCarbohidratos, GlucogénesistipoI .DeficienciaGLUCOSA6FOSFATASA AutosómicaRecesiva Tamizmetabólico Tx.almidón . AfectaciónHEPÁTICA . Hepatomegalia . Hipoglucemia Factoresderiesgo: Consanguinidad Williams,Síndromede Cromosomopatíaestructural,Síndromedegenes contiguos (MICRODELECIÓN7q11) Haploinsuficienciadel gen p/Elastina FISH . Déficitintelectual . Hipercalciemia(infancia) . Labioinferiorgrueso . Hombroscaídos . Filtrum largo . Trastornosdelosgrandesvasos (Estenosis Aortica) . Extrovertidos de niños,introvertidos de adulto (Música) Wolf-Hirschhorn,Síndromede Cromosomopatíaestructural, (DELECIÓN4p16.3) Mutacióndenovo 87% Progenitorc/translocación 13% Ultrasonido Cariotipo FISH . Retrasomentalgrave . Convulsionesmioclónicas . Hipotonía . Microcefalia . Aspecto facialdeCASCO GRIEGO . Hendiduraspalpebralesinclinadashaciaabajo . Hipertelorismo . Frenteamplia X frágil,Síndromede Trastornodelneurodesarrollo CromosomaXq21.3,Gen FMR1,locusFRAXA ExpansióntripletesCGG (entre 59-199:PREMUTACIÓN; arribade 2OO:MUTACIÓN) PCR metilada Mayorfrecuenciaenhombres . Orejasgrandes . Articulacioneshiperextensibles . Prolapsodela válvula mitral Factoresderiesgo: Sexo masculino