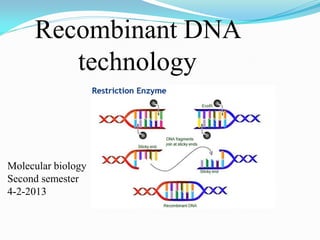
2. A series of procedures used to recombine DNA
segments. Under certain conditions, a
recombinant DNA molecule can enter a cell and
replicate.
3. Recombinant DNA technology is one of the
recent advances in biotechnology, which was
developed by two scientists named Boyer and
Cohen in 1973.
4. RECOMBINANT DNA
RECOMBINANT DNA:-
DNA molecules constructed outside of living cells by
joining natural or synthetic DNA segments to DNA
molecules that can replicate in a living cell
5. The DNA is inserted into another DNA
molecule called ‘vector’
The recombinant vector is then introduced
into a host cell where it replicates itself, the
gene is then produced
7. How is Recombinant DNA
made?
There are three different methods by
which Recombinant DNA is made.
They are Transformation, Phage
Introduction, and Non-Bacterial
Transformation.
8. Transformation
The first step in transformation is to select a piece of
DNA to be inserted into a vector. The second step is to
cut that piece of DNA with a restriction enzyme and then
ligate the DNA insert into the vector with DNA Ligase.
The insert contains a selectable marker which allows for
identification of recombinant molecules. The vector is
inserted into a host cell, in a process called
transformation. One example of a possible host cell is E.
coli. The host cells must be specially prepared to take up
the foreign DNA.
9. Non-Bacterial transformation
Microinjection, the DNA is injected
directly into the nucleus of the cell being
transformed. The host cells are bombarded
with high velocity micro-projectiles, such
as particles of gold or tungsten that have
been coated with DNA.
10. Phage Introduction
Phage introduction is the process of transfection,
which is equivalent to transformation,
except a phage is used instead of bacteria. In vitro
packaging of a vector is used. This uses lambda or
MI3 phages to produce phage plaques which
contain recombinants. The recombinants that are
created can be identified by differences in the
recombinants and non-recombinants using various
selection methods.
11. How does rDNA work?
Recombinant DNA works when the host cell expresses
protein from the recombinant genes. A significant
amount of recombinant protein will not be produced
by the host unless expression factors are added.
Protein expression depends upon the gene being
surrounded by a collection of signals which provide
instructions for the transcription and translation of the
gene by the cell. These signals include the promoter,
the ribosome binding site, and the terminator.
12. Expression vectors, in which the foreign DNA is
inserted, contain these signals. Signals are species
specific. In the case of E. coli, these signals must be E.
coli signals as E. coli is unlikely to understand the
signals of human promoters and terminators.
Problems are encountered if the gene contains introns
or contains signals which act as terminators to a
bacterial host. This results in premature termination,
and the recombinant protein may not be processed
correctly, be folded correctly, or may even be
degraded.
13. Production of recombinant proteins in
eukaryotic systems generally takes place in
yeast and filamentous fungi. The uses of
animal cells is difficult due to the fact that
many need a solid support surface, unlike
bacteria, and have complex growth needs.
However, some proteins are too complex
to be produced in bacterium, so eukaryotic
cells must be used.
14. Large-scale production of human proteins
by genetically engineered bacteria.
Such as : insulin, Growth hormone,
Interferons and
Blood clotting factors (VIII & IX)
15. 1) Obtaining the human insulin gene
Human insulin gene can be obtained by making a
complementary DNA (cDNA) copy of the messenger
RNA (mRNA) for human insulin.
16. 2)Joining the human insulin gene
into a plasmid vector
The bacterial plasmids and the cDNA are
mixed together. The human insulin gene
(cDNA) is inserted into the plasmid through
complementary base pairing at sticky ends.
17. 3)Introducing the recombinant
DNA plasmids into bacteria
The bacteria E.coli is used as the host cell. If E.
coli and the recombinant plasmids are mixed
together in a test-tube.
18. 4)Selecting the bacteria which
have taken up the correct
piece of DNA
The bacteria are spread onto nutrient agar. The
agar also contains substances such as an
antibiotic which allows growth of only the
transformed bacteria.
19. Vaccine development
The surface antigen of Plasmodium
falciparum, one of the 4 species of malaria,
has been transferred to E. coli to produce
amounts large enough to develop a vaccine
against this form of malaria. It works well
enough for people who will visit a
malarious region for a relatively short
period of time.
20. Hemophilia A and B
The genes encoding factors 8 and 9 are on the X
chromosome.
Like other X-linked disorders, hemophilia A and B are found
almost exclusively in males because they inherit just a
single X chromosome, and if the gene for factor 8 (or 9) on
it is defective, they will suffer from the disease.
There are many different mutant versions of the genes for
factors 8 and 9. Although some produce only a minor effect
on the function of their protein, others fail to produce any
functioning clotting factor.
21. Transferring the gene for normal adult
hemoglobin into marrow stem cells of an
individual with sickle-cell anemia. The goal
is to promote the growth of enough cells to
produce enough normal hemoglobin to
alleviate the symptoms of sickle-cell anemia.
Gene therapy for genetic diseases
22. Safety Issues in relation to Recombinant
DNA Technology
As bacteria is commonly used in recombinant DNA work,
there has always been a concern among scientists and a
worry among people that there is a possibility that a clone
of highly pathogenic recombinant bacteria were made by
accident, then escaped from the laboratory and caused an
epidemic for which no drugs were available.
Recombinant DNA Advisory Committee (RAC) was
established in 1974 in the United States, which
responds to public concerns regarding the safety of
manipulation of genetic material through the use of
recombinant DNA techniques.
23. 2 types of control : physical
containment and biological
containment
Effective biological safety programs were
operated in a variety of laboratories, which
include a set of standard practices generally
used in microbiological laboratories, and
special procedures, equipment and laboratory
installations that provide physical barriers of
varying degrees.
24. In considering biological containment, the
vector (plasmid, organelle, or virus) for the
recombinant DNA and the host (bacterial,
plant, or animal cell) in which the vector is
propagated in the laboratory will be considered
together.
(i) survival of the vector in its host outside the
laboratory, and (ii) transmission of the vector
from the propagation host to other non-
laboratory hosts.
Biological containment
25. It is always possible that an antibiotic-resistant
plasmid could be accidentally incorporated into
a dangerous pathogen with serious medical
consequences.
27. Introduction
Within living cells, the exchange of DNA sequences and
genetic information can occur through a regulated series
enzymatic reactions involving pairing of DNA molecules
and phosphodiester bond breakage and rejoining. This
type of sequence rearrangement is known as genetic
recombination. genetic recombination responsible for
rearranging sequences between different pieces of DNA,
shaping the genome by altering the sequences that are
present, pairing chromosome before cell division and
promoting DNA repair.
29. Biological Roles for Recombination
1. Generating new gene/allele combinations
(crossing over during meiosis)
2. Generating new genes (e.g., Immuno- globulin
rearrangement)
3. Integration of a specific DNA element
4. DNA repair
30. Practical Uses of Recombination
1. Used to map genes on chromosomes
(recombination frequency proportional to
distance between genes)
2. Making transgenic cells and organisms
31. Map of Chromosome I of
Chlamydomonas reinhardtii
cM = centiMorgan; unit of recombination frequency
1 cM = 1% recombination frequency
32. Types of Recombination
1. Homologous - occurs between sequences
that are nearly identical (e.g., during
meiosis)
2. Site-Specific - occurs between sequences
with a limited stretch of similarity; involves
specific sites
3. Transposition – DNA element moves from
one site to another, usually little sequence
similarity involved
33. Homologous recombination
Is the exchange of DNA sequences between DNA
molecules that contain identical or nearly identical
sequences along their length.
The common stretch of bases that will be recombined
is known as the homology between the sequences and
can be as few as 50-100bp or as much as a whole
chromosome.
The greater the region of homology the higher the
frequency of recombination.
34. Requirement of homologous recombination
1- Two DNA sequences with similar or identical base
pair sequences.
2- The ability to form stable hydrogen bonds between
the bases on one strand of one DNA sequence and the
base on the complementary strand on the other DNA
sequence.
3- The proteins needed to carry out recombination.
35. Rec A, the most important protein in DNA
recombination.
38 kDa protein that polymerizes onto SS DNA 5’-3’,
Catalyzes strand exchange, has an ATPase,
Also binds DS DNA, but not as strongly as SS
36. RecA, mechanism of action
3 steps of strand exchange:
1. Pre-synapsis: recA coats single stranded
DNA (accelerated by SSB, get more relaxed
structure)
2. Synapsis: alignment of complementary
sequences in SS and DS DNA (paranemic
or side-by-side structure)
3. Post-synapsis or strand-exchange: SS DNA
replaces the same strand in the duplex to
form a new DS DNA (requires ATP
hydrolysis)
37.
38. But what create a single strand
DNA in the cell?
Non enzymatic (physical agents)
Enzymatic (specific enzyme responsible for creating
ssDNA). This enzyme is known as RecBCD.
39. RecBCD : A complex enzyme
RecBCD has several enzymatic function:
1. Endonuclease subunits (recBCD) that cut one
DNA strand close to Chi sequence.
2. DNA helicase activity (recBC subunit) and
3. DNA-dependent ATPase activity
unwinds DNA to generate SS regions
40.
41. recBCD Pathway of Homologous Recombination
Part I: Nicking and Exchanging
1. A nick is created in one strand by recBCD at a Chi
sequence (GCTGGTGG), found every 5000 bp.
2. Unwinding of DNA containing Chi sequence by
recBCD allows binding of SSB and recA.
3. recA promotes strand invasion into homologous
DNA, displacing one strand.
4. The displaced strand base-pairs with the single
strand left behind on the other chromosome.
5. The displaced and now paired strand is nicked
(by recBCD?) to complete strand exchange.
45. RuvA and RuvB
DNA helicase that catalyzes branch migration
RuvA tetramer binds to HJ (each DNA helix
between subunits)
RuvB is a hexamer ring, has helicase & ATPase
activity
2 copies of ruvB bind at the HJ (to ruvA and 2 of
the DNA helices)
Branch migration is in the direction of recA
mediated strand-exchange
46.
47. RuvC : resolvase
Endonuclease that cuts 2 strands of HJ
Binds to HJ as a dimer.
Consensus sequence: (A/T)TT (G/C)
- occurs frequently in E. coli genome
- branch migration needed to reach
consensus sequence!
49. Models of recombination:
1- copy choice model:
• In this model replication is a requirement for
recombination (DNA pol III switches positions,
alternating back and forth of between two DNA
molecules acting as replication template).
50.
51. 2- Holliday Model
R. Holliday (1964)
- Holliday Junctions
form during
recombination
- HJs can be resolved
2 ways, only one
produces true
recombinant
molecules
52. EM of a Holliday Junction w/a few melted
base pairs around junction
54. DNA exchange in bacteria
Plasmids
Methods of DNA
2nd semeste
20-2-2012
55. Introduction
Bacteria can exchange or transfer DNA between other
bacteria in three different ways. In every case the
source cells of the DNA are called the DONORS and
the cells that receive the DNA are called
the RECIPIENTS. In each case the donor DNA is
incorporated into the recipients cell's DNA by
recombination exchange.
55
56. Plasmids are pieces of DNA
that exist separate from the
chromosome. They contain
an Origin of replication
(ori) and, as such Replicate
independentely from the
chromosome.
Plasmid contain a variety
other genes; antibiotic
resistant gene, degradation
of different carbon sources
and genes involved in
causing disease
56
Plasmids
57. DNA synthesis from
the origin proceeding
in one direction at a
time (unidirectional
DNA synthesis) or
both directions
(bidirectional DNA
synthesis).
57
58. Replicon
Because plasmids have a mechanism to
replicate their DNA and ensure that they
are stably maintained in a cell population,
they are also known as replicon.
58
59. Plasmid copy number
Low copy number plasmids (1-2 copy): P1 phage & F
plasmid or pSC101 (10-15 copy)
High copy number plasmids (50 copies): ColE1 (50)
59
60. Plasmid partitioning
High copy no. plasmid
do not have any
mechanism of
partitioning .
Low copy no.
plasmids must have a
mechanism to ensure
their proper
partitioning.
60
61. parS : A specific site on low copy number plasmids
required for segregation.
ParA & parB proteins bind to parS sequence,
61
62. How parS, parA and parB protein
complex function?
Keep the plasmid molecule in the middle of the cell until
daughter cell are clearly distinguished.
How the plasmids distinguished between the different
physical locations in the cell?
62
64. Plasmid incompatibility
Maintenance of more than one plasmids in the cell can
only occur if the plasmids can only carry different
origins of replication.
The inability of two plasmids with the same origins to
be maintained in the same cell is known as
incompatibility.
64
66. Incompatibility can be explained by a
limiting concentration of initiator
protein and random replication of
plasmid molecules. Plasmid molecules
are randomly chosen from the
population and undergo DNA
replication each time the plasmid is
replicated. Different plasmids that use
the same or very similar origins and
initiator proteins are rapidly separated
from each other.
66
67. Broad host range plasmids
Plasmid that can be replicated and maintained in
many different bacterial species are known as broad
host range plasmids.
67
68. Moving plasmids from one cell to
another.
By
* Conjugation
(Shuttle vector)
Risk:
Moving antibiotics resistance between bacterial species.
68
69. Gene exchangeRequirement Contact Genes transferred
in most
Transformation
Free donor DNA
fragment
Competent recipient
cell
no
Most capsule gene,
unlimited
Conjugation
Sex pili on donor
Fertility plasmid in
donor
Live cells
Closely related species
yes Drug resistance,
resistant genes
to toxin,
enzymes etc.
Transduction
Donor lysed by phage
Defective phage
carries donor DNA
Live competent
recipient cell
no
Toxins, drug
resistance
70. Bacterial conjugation is the
transfer of genetic material
between bacteria through
direct cell to cell contact, or
through a bridge-like
connection between the two
cells. conjugation is a
mechanism of horizontal
gene transfer.
71. The prototype for conjugative plasmids is the F-
plasmid, also called the F-factor.
The F-plasmid is an episome (a plasmid that can
integrate itself into the bacterial chromosome by
homologous recombination) of about 100 kb length.
It carries its own origin of replication, the oriV, as well
as an origin of transfer, or oriT.
There can only be one copy of the F-plasmid in a given
bacterium, either free or integrated.
The host bacterium is called F-positive or F-plus
(denoted F+). Strains that lack F plasmids are called F-
negative or F-minus (F-).
72. Among other genetic information, the F-plasmid
carries a tra and a trb locus, which together are about
33 kb long and consist of about 40 genes.
The tra locus includes the pilin gene and regulatory
genes, which together form pili on the cell surface,
polymeric proteins that can attach themselves to the
surface of F- bacteria and initiate the conjugation.
74. When conjugation is initiated, a relaxase enzyme
creates a nick in one plasmid DNA strand at the origin
of transfer, or oriT.
The relaxase may work alone or in a complex of over a
dozen proteins, known collectively as a relaxosome.
In the F-plasmid system, the relaxase enzyme is called
TraI and the relaxosome consists of TraI, TraY, TraM,
and the integrated host factor, IHF.
75. The transferred, or T-strand, is unwound from the
duplex plasmid and transferred into the recipient
bacterium in a 5'-terminus to 3'-terminus direction.
The remaining strand is replicated, either independent
of conjugative action beginning at the oriV) or in
concert with conjugation (conjugative replication
similar to the rolling circle replication of lambda
phage).
76.
77. If the F-plasmid becomes integrated into the host
genome, donor chromosomal DNA may be transferred
along with plasmid DNA and the cell is called Hfr.
The certain amount of chromosomal DNA that is
transferred depends on how long the bacteria remain
in contact; for common laboratory strains of E. coli the
transfer of the entire bacterial chromosome takes
about 100 minutes. The transferred DNA can be
integrated into the recipient genome via homologous
recombination.
78. Some strains of bacteria with an integrated F-plasmid
can be isolated and grown in pure culture. Because
such strains transfer chromosomal genes very
efficiently, they are called Hfr (high frequency of
recombination).
78
79. Formation of the Hfr
F plasmid recombine to the chromosome by
homologous recombination.
F plasmid carry two copies of IS3, one copy of IS2 and
one copy of Tn1000.
The chromosome has about six copies of IS2, five
copies of IS3 and multiple copies of Tn1000.
These sites can be used as a source of homologous
recombination.
79
80. Formation of the F´( F prime)
F´ are formed from Hfr strains.
The F plasmid that is integrated into the chromosome
can come back.
The F factor can come back out with only F plasmid
reforming F+.
The F factor can come back out and carry
chromosomal DNA either from left or right of the
original insertion these are known as type I F prime.
80
81. If the F´ carry genes that were transferred early by the
starting Hfr strain then it’s a Type IA F´,
If the F´ carry genes that were transferred late by the
starting Hfr strain then it’s a
Type IB F´,
Its possible for the F factor to carry genes from both
sides of the original chromosome and these are called
Type IIF´
81
82. Genetic uses of F´
Merodiploid ( two copies of the gene into the same
cell)
82
85. Transformation: Is the process that allows bacteria to take
up free or naked DNA from their surrounding
environment.
Bacteria that have undergoes transformation are called
transformants.
Transformation requires that the bacterium can become
naturally competent to take up DNA from their
surrounding environment.
86. Natural competency
When do bacterial cell become competent to take up
DNA ?
the physiological state of the cell influences its ability
to become competent.
87. Establishment of the competent
cell Based on
Cell density (end of the exponential phase)
Depletion carbon, nitrogen and phosphorus
Transformation Sporulation
88. Two pathways activate the
competence genes in B.
subtilis. P1 sense the cell
density, and when enough
cells are present, signals
the cells to induce the com
genes. P2 monitors the
available nutrients and
when they become
limiting, signals the cells to
induce the com genes.
89. One pathway used by B. subtillis relies on a small peptide,
com X to sense the cell density and induce the com genes.
ComX is produced in the cytoplasm as a larger precursor
molecule, the comQ protein processes and modifies
precursor comX to make a smaller active peptide. The
active comx is excreted from the cell.
90. Two component signal
transduction systems use a
membrane protein called
sensor-kinase to sense an
environmental signal (a). The
sensor - kinase
autophosphorylates on a
specific histidine residue in
the protein(b). The sensor-
kinase transfer this
phosphate to the response
regulator (c). This activates
the response regulator so that
it can induce and/ or repress
specific genes (d). These
genes allow a cell to respond
to the original environmental
signal in an appropriate way.
91. ComP and comA comprise a
two components signal
transduction system in B.
subtillis for the induction of the
competency genes. (a) ComX
binds to comP. (b) This binding
induce autophosphorylation of
comP. (c) The phosphate is
relied to comA. (d) the
phosphorylated comA activates
the comS genes. (e) ComS leads
to the induction of the
competency genes.
92. Quorum sensing system
ComP, comA and comX depend on cell density
therefore they called quorum sensing system.
93. The second pathway to
induce the competency
genes in B.subtillis uses
another small peptide
called CSF.
CSF transported to the
cell via membrane
protein called spo0k .
94. The process of
transformation
Transformation in B. subtillis
a- A type IV pillus binds the
double stranded DNA first.
b- The DNA is transferred to
the comEA receptor.
c- An endonuclease comI
recognizes the receptor with
bound DNA.
d- ComI degrade one strand of
the DNA.
e- The single stranded DNA is
transported in to the cell and
protected by a single
stranded DNA binding
protein.
95. Some species that are
naturally competent can
only take up DNA that
contains specific base
sequence called USS or
uptake signal sequence.
96. Some species used a
specialized structure
called a transformasome
to import DNA. The
DNA most contain a USS
and initially is double
stranded. During the
process of translocation,
the DNA is degraded to a
single – stranded
molecule.
97. Transformation as a genetic tool:
gene mapping
Mapping genes requires that a piece of chromosomal
DNA from one bacterium be introduced to another
bacterium of the same species.
Transformation as a molecular tool:
Introducing genes to many different bacteria.
99. Aim of this lecture
To understand the method of moving DNA by phage
To understand generalized and specialized transducing
phage
To differentiate between screening and selection.
To know the benefit of transduction.
100. Transduction:
Is the process of moving a piece of chromosome (DNA )
from one cell to another using a bacteriophage to carry
DNA.
First described by Zinder and Lederburg in 1952.
101. There are two types of transducing phage
1- Generalized: capable of moving any piece of DNA from
one cell to another.
2- specialized: move the same piece of DNA every time.
102.
103. P1 as a model for generalized
transducing phage
P1 has a double stranded DNA (90kb).
P1 infect E.coli and package DNA into a phage head
from a specific site on the P1 DNA known as pac site,
once the DNA is inserted into the phage head, a p1tail is
added to make a complete phage that is capable of
infecting another E.coli cell.
104. Specialized transducing phage
Specialized transducing phage carrying a defined region
of the chromosome.
Unlike generalized transduing phage specialized
transducing phage carry chromsomal DNA in addition to
phage DNA.
Example is λ.
105. λ recombine with the bacterial chromosome using attB
site by site specific recombination.
106. Identifying transdused bacteria
Screening: examining each individual bacterium is
known as screening.
Selection: bacteria with specific characteristics or
phenotype can be selected by growth or non growth
on specified media.
A- Lethal selection
B- non lethal selection
107. Uses for transduction
1- Two factor crosses to determine gene linkage.
2- Mapping the orders of the gene – three factor crosses.
3- strain construction
4- Localized mutagenesis.
5- Making merodiploides using specialized transduction.
6- Moving mutations from plasmids to specialized
transducing phage to the chromosome.
108. DNA Cloning
This lecture is concerned with the details of the individual
steps in the cloning process:
1- How is the DNA removed from the cell?
2- How is the DNA is cut into pieces?
3- How are the pieces of DNA put back together?
4- How we do monitor each of these steps?
109. DNA Cloning
Isolation DNA from cells.
Plasmid DNA isolation.
Chromosomal DNA isolation.
Cutting DNA molecules (Restriction enzymes).
Joining DNA molecules.
Application of DNA cloning (DNA library).
DNA detection
1- Southern blotting
2- DNA amplification PCR.
110. Plasmid DNA isolation
The first step in DNA cloning is isolating of the vector
and chromosomal DNA.
In the general scheme, cells containing the plasmid are
grown to a high cell density, gently lysed, and the plasmid
DNA is isolated and concentrated.
111. Cells can be lysed by different method depending on the
size of plasmid molecule.
Most procedures used EDTA to chelate the Mg+ ²
associated the outer membrane and destabilize the outer
membrane.
Lysosyme is added to digest the peptidoglycan and
detergent are frequently used to solubilize the membrane.
RNAses are added to degrade the large amount of RNA
found in the actively growing cells.
(for more detail see the practical part)**
112. Chromosomal DNA isolation
To isolate chromosomal DNA, cells are lysed in the same
way as for plasmid DNA isolation.
The cell lysate is extracted using phenol or otherwise
treated to remove all proteins.
The chromosomal DNA is very fragile and breaks easily,
for this reason, the chromosomal DNA is not usually
purified using columns.
113. Cutting DNA Molecule
Once DNA has been purified, it must be cut into pieces
before the chromosomal DNA and the plasmid DNA can
be joined.
A group of enzymes called restriction enzymes are used
for cutting DNA.
114. Why bacteria use restriction and modification enzyme?
Bacteria use restriction and modification enzymes to
identify their own DNA from any foreign DNA that enter
their cytoplasm.
The restriction part of the system is an enzyme that
recognize a specific site on DNA called restriction site and
cleaves DNA by catalyzing breaks in specific
phosphodiester bonds on both strands of DNA.
The modification part of the system is a protein that
methylates the DNA sequence so that the restriction
enzyme no longer recognize the DNA sequence. Thus the
bacteria can protect its own DNA from the restriction
enzyme.
115. Types of restriction enzymes
1- Type I restriction–modification systems
Three different proteins form a complex that carries out both
restriction and modification of the DNA. The complex must
interact with a cofactor, S- adenosylmethionine, before it is
capable of recognizing DNA.
The S-adenosylmethionine is the methyl donor for the
modification reaction and all known Type I systems methylate
adenine residues on both strands of the DNA.
The restriction reaction requires ATP and Mg++ for cleavage
of the DNA.
The DNA sequence recognized by Type I enzymes is
asymmetric. Cleavage of the DNA occurs randomly, usually no
closer than 400 bp from the recognition sequence and
sometimes as far away as 7000 bp.
116. Type II restriction–modification systems
Composed of two independent proteins. One protein is
responsible for modifying the DNA and one for restricting the
DNA.
Modification of the DNA uses S- adenosylmethionine as the
methyl donor.
The modifications that have been found are 5-methlycytosine,
4-methylcytosine, or 6-methlyadenosine.
The DNA sequence recognized by Type II restriction enzymes
is symmetric and usually palindromic.
Both the cleavage of the DNA and modification of the DNA
occur symmetrically on both strands of the DNA within the
recognition sequence.
Several thousand Type II systems have been identified. Type II
restriction enzymes are the most useful for cloning
because they generate DNA molecules with a specific sequence
on the ends.
117. Type III restriction–modification systems
Type III systems are composed of two different proteins in a
complex. The complex is responsible for both restriction and
modification.
Modification requires S-adenosylmethionine, is stimulated by
ATP and Mg++, and occurs as 6-methyladenine.
Restriction requires Mg++ and is stimulated by ATP and S-
adenosylmethionine.
The recognition sites for Type III enzymes are asymmetric
and 5–6 bp in length.
The DNA is cleaved on the 3´side of the recognition sequence,
25–27 bp away from the recognition sequence.
Type III restriction enzymes require two recognition sites in
inverted orientation in order to cleave the DNA.
119. Type II restriction enzymes generate DNA molecules with specific sequences
on both ends. These ends can be rejoined to regenerate the restriction site.
120. Restriction–modification as a molecular tool
The cleavage is on both strands of the DNA and results in
a double- stranded break. Cleavage of the DNA leaves one
of three types of ends, depending upon the specific
restriction enzyme.
Some enzymes leave a 5´ overhang, some a 3´ overhang,
and some leave blunt ends. The ends with either a 5´ or 3 ´
overhang are known as sticky ends.
121.
122. Joining DNA molecules
The double-stranded ends must be covalently attached. A version of
this reaction is normally carried out in the cell by an enzyme known
as DNA ligase.
The double-stranded break formed by the restriction enzyme can be
thought of as two nicks, each of which is a substrate for ligase.
If a plasmid molecule that has been digested with a restriction
enzyme is subsequently treated with ligase, the plasmid molecule
ends can be covalently closed by ligase .
Ligation is an energy-requiring reaction that occurs in three distinct
steps.
1- The adenylyl group from ATP is covalently attached to ligase and
inorganic phosphate is released.
2- The adenylyl group is transferred from ligase to the 5´ phosphate of
the DNA in the nick.
3- The phosphodiester bond is formed when the 3 ´ OH in the nick
attacks the activated 5 ´ phosphate. AMP is released in the process.
125. Aims of this lecture
1- To describe how a library of DNA
can be constructed using DNA
cloning method.
2- To understand how cloned DNA
can be detected by southern blotting
and PCR.
126. Introduction In 1962 the Noble prize in medicine and physiology was
awarded to Watson and Crick for the discovery of the
structure of DNA. The technology developed in the 50
years since has revolutionized how biological research is
conducted. The ability to manipulate genes in vitro has
greatly increased not only the experiments that are know
possible but also how scientists think about biological
problems. Each of the techniques that will be describe in
this lecture allows scientists to manipulate a novel gene in
many different ways with the goal of uncovering its
unique role in the cell.
128. DNA libraries –a collections of DNA sequences.
DNA libraries, like conventional libraries, are used to
collect and store information.
In DNA libraries, the information is stored as a set of
DNA molecules.
All DNA libraries are collections of DNA fragments that
represent a particular biological system of interest.
The two most common uses for these DNA collections
are DNA sequencing and gene cloning.
129. A DNA library is a collection of clones of DNA designed
so that there is a high probability of finding any particular
piece of the source DNA in the collection.
DNA libraries can be made using highly efficient cloning
vectors such as lambda phages, plasmids, cosmids, P1
phages and bacterial or yeast artificial chromosomes.
130. Types of DNA Libraries
The genomic library contains DNA fragments
representing the entire genome of an organism.
The cDNA library contains only complementary DNA
molecules synthesized from mRNA molecules in a cell.
131. Genomic Library
Are made from total nuclear DNA of an organism or
species.
DNA is cut into clonable size pieces as randomly as
possible using restriction endonuclease.
Genomic libraries contain whole genomic fragments
including gene exons and introns, gene promoters,
intragenic DNA, centromeric DNA, origins of replication,
etc
132. Constructing libraries of clones
The library is made by inserting these millions of
fragments of DNA into λ bacteriophage plasmids.
This allows the genes to be grown up (cloned) in E. coli.
The library can be screened for DNA fragments or
Particular genes
133.
134. cDNA Library
The advantage of cDNA library is that it contains only the
coding region of a genome.
135.
136. Vectors for DNA Libraries
Genomic libraries
– λ-phage - 9-23 kb → convenient and easy to handle
– Cosmids - 30-45
– PAC, BAC, YAC →artificial chromosomes,
accommodate large fragments
cDNA libraries
-λ-phage - 9-23 kb → provides selection for longer
cDNAs
-conventional plasmids → high level of expression of
proteins.
137. DNA detection—Southern
blottingIn 1975, E.M. Southern described a technique
to detect sequence homology between two
molecules, without determining the exact base
sequence of the molecules.
The Southern blot is used to detect the
presence of a particular piece of DNA in a
sample.
The DNA detected can be a single gene, or it
can be part of a larger piece of DNA such as a
viral genome.
138. The key to this method is hybridization.
Hybridization-process of forming a double-stranded DNA
molecule between a single-stranded DNA probe and a
single-stranded target patient DNA.
139. There are 2 important features of hybridization:
The reactions are specific-the probes will
only bind to targets with a complementary
sequence.
The probe can find one molecule of target in
a mixture of millions of related but non-
complementary molecules.
140. Steps for hybridization
The technique relies on fractionating the DNA
on an agarose gel and denaturing the
fractionated DNA in the agarose. The
denatured DNA is transferred to a solid
support, such as a nylon or nitrocellulose
filter.
A second DNA, called the probe, is labeled
with a tag, denatured, and applied to the filter.
Probes can be tagged with radioactivity and
detected with X-ray film.
141. They can also be labeled with fluorescent nucleotides or
enzymes such as alkaline phosphatase or horseradish
peroxidase.
The enzymes are then detected with special substrate
molecules that change color or emit light when cleaved by
the enzyme.
The probe will hybridize with any DNA on the filter that
has complementary base sequences. Once the excess, non-
hybridized probe is washed away, the tag attached to the
probe can be detected.
142. Definition of DNA probe :
a single-stranded DNA molecule used
in laboratory experiments to detect
the presence of a complementary
sequence among a mixture of other
singled-stranded DNA molecules.
144. RNA blot : Northen Blot
It can be used to determine the temporal and spatial
locations of RNA expression by ―running‖ an RNA blot,
often referred to as a northern blot.
148. USES
Identify mutations, deletions, and gene
rearrangements.
Used in diagnosis of cancer and in
prenatal diagnosis of genetic diseases
Leukemias.
Diagnosis of HIV-1 and infectious
disease.
149. USES
Every person has repeated sequences of
base pairs which are called Variable
Number Tandem Repeats (VNTRs)
To find a particular VNTR we use a
radioactive version of the one in question.
This pattern is known as a DNA
fingerprint.
150. USES
Applications of DNA fingerprinting
include:
Paternity and Maternity Testing
Criminal Identification and
Forensics
Personal Identification
151. DNA sequencing
Introduction:
Knowledge of DNA sequences has become indispensable for
basic biological research, other research branches utilizing
DNA sequencing, and in numerous applied fields such as
diagnostic, biotechnology, forensic biology and biological
systematics. The advent of DNA sequencing has significantly
accelerated biological research and discovery. The rapid speed
of sequencing attained with modern DNA sequencing
technology has been instrumental in the sequencing of the
human genome, in the human genome project. Related projects,
often by scientific collaboration across continents, have
generated the complete DNA sequences of many animal, plant,
and microbial genomes.
152. DNA sequencing includes several methods and
technologies that are used for determining the order of the
nucleotide bases—adenine, guanine, cytosine, and
thymine—in a molecule of DNA.
153. Two similar methods used for determining the order of the nucleotide
sequences:
1. Maxam and Gilbert method
2. Sanger method
They depend on the production of a mixture of oligonucleotides labeled
either radioactively or fluorescing, with one common end and differing in
length by a single nucleotide at the other end
This mixture of oligonucleotides is separated by high resolution
electrophoresis on polyacrilamide gels and the position of the bands
determined
154. The Maxam-Gilbert
Technique
Principle - Chemical Degradation of
Purines
Purines (A, G) damaged by
dimethylsulfate
Methylation of base
Heat releases base
Alkali cleaves G
Dilute acid cleave A>G
156. Maxam and Gilbert Method
Chemical degradation of purified fragments (chemical degradation)
The single stranded DNA fragment to be sequenced is end-labeled by
treatment with alkaline phosphatase to remove the 5’phosphate
It is then followed by reaction with P-labeled ATP in the presence of
polynucleotide kinase, which attaches P labeled to the 5’terminal
The labeled DNA fragment is then divided into four aliquots, each of which is
treated with a reagent which modifies a specific base
1. Aliquot A + dimethyl sulphate, which methylates guanine residue
2. Aliquot B + formic acid, which modifies adenine and guanine residues
3. Aliquot C + Hydrazine, which modifies thymine + cytosine residues
4. Aliquot D + Hydrazine + 5 mol/l NaCl, which makes the reaction specific for cytosine
The four are incubated with piperidine which cleaves the sugar phosphate
backbone of DNA next to the residue that has been modified
158. Chain-termination methods (Sanger method)
The key principle of the Sanger method was the use of
dideoxynucleotide triphosphates (ddNTPs) as DNA chain
terminators.
The classical chain-termination method requires
1- single-stranded DNA template
2- DNA primer
3- DNA polymerase
4- normal deoxynucleotidephosphates (dNTPs)
5-modified nucleotides (dideoxyNTPs) that terminate DNA
strand elongation.
These ddNTPs will also be radioactively or fluorescently
labeled for detection in automated sequencing machines.
159. In a synthesis reaction, if a dideoxynucleotide
is added instead of the normal
deoxynucleotide, the synthesis stops at that
point because the 3’OH necessary for the
addition of the next nucleotide is absent.
160. In the dideoxy method of sequencing, the template DNA that is to be
sequenced is mixed with a primer complementary to the template DNA
and the four normal dNTPs, one of which is radioactively labeled for
subsequent visualization purposes.
This mixture is then splint into four different tubes that are labeled A, C,
G, and T. Each tube is then ―spiked‖ with a different ddNTP (ddATP for
tube A, ddCTP for tube C, ddGTT for tube G, or ddTTP for tube T).
DNA polymerase is added and using the DNA template and its’
complementary primer, the synthesis of new strands of DNA
complementary to the template begins.
Occasionally a dideoxynucleotide is added instead of the normal
deoxynucleotide and synthesis of that strand is terminated at that point.
161. In the tube containing ddATP, some percentage of newly synthesized
molecules will get a ddATP in each place that there is a T in the template
DNA.
The result is a set of new DNA molecules in tube A, each of which ends
in an A.
A similar type of reaction occurs in the three other tubes to result in
molecules that end in C, G, and T in tubes C, G, and T respectively.
After the synthesis reactions are complete, the products of the four
different tubes are loaded onto four adjacent lane of a polyacrylamide gel
and the different fragments are separated by size.
The sequencing gel is able to resolve fragments that differ in size from
each other by only one base.
162. After electrophoresis to separate the fragments by size,
the fragments are visualized to exposing the gel to
photographic film (Remember that one nucleotide was
radioactively labeled).
All fragments in lane A will end in an A, fragments in
lane C will all end in a C, fragments in lane G will all
end in a G, and fragments in lane T will all end in a T.
The sequence of the DNA is read from the gel by
starting at the bottom and reading upward.
167. Dye-terminator sequencing
Automated DNA sequencing – in automated DNA
sequencing a radioactive deoxynucleotide is not used and all
four dideoxy reactions are done in a single tube.
This is possible because each ddNTPs is labeled with a
different flourescent dye.
Therefore the dye present in each synthesized fragment
corresponds to the dye attached to the dideoxynucleotide
that was added to terminate the synthesis of that particular
fragment.
The contents of the single tube reaction are loaded onto a
single lane of a gel and electrophoresis is done.
168. A flourimeter and computer are hooked up to the gel
and they detect and record the dye attached to the
fragments as they come off the gel.
The sequence is determined by the order of the dyes
coming off the gel.
172. Automated DNA Sequencing with Fluorescent Dyes
Each different ddNTP is coupled to a different colored fluorescent dye
ddTTP is red; ddGTP is black etc.